Cystic Fibrosis can impact many parts of the body. Continue exploring this section to learn more about the various systems and how they might be affected.
Basic Facts
Cystic fibrosis (CF) is a common genetic disease that is inherited from your biological parents. It is the most common genetic disease in Caucasians; approximately 1 in every 3,600 children born in Canada has CF.
Genetic illnesses occur because of abnormalities in our genes.
All the cells in our body contain DNA. Simply put, our DNA is the instructions on how to build us. If you think of our DNA like a library, then all these instructions are divided into “books” – called chromosomes and within each book, there are “chapters” called genes. Each gene contains detailed instructions on how to build one part of our body.
Just like there can be mistakes in written instructions, there can be mistakes in our DNA. A mistake in our DNA is called a mutation.
The CF gene is the instructions on how to build a channel (or pore) in our cells that allows salts (chloride and bicarbonate) to move into and out of the cell. This salt channel is called CFTR, the cystic fibrosis transmembrane regulator. A mutation in the CF gene means that there is a mistake in the instructions on how to build this salt channel so it does not work properly.
CFTR is important in regulating the amount of water in the mucus in our bodies – for example in the lungs, sinuses, and digestive tract. If CFTR does not work properly, the mucus is abnormally thick and sticky.
We inherit a copy of our genes from our parents – one copy from our father and one from our mother. If only one copy of the CF gene with a mistake (mutation) is inherited, then the individual is a “carrier” of the CF gene. This is because they carry the mutation (and can pass it on to their children), but because the copy of the gene inherited from the other parent is normal, the salt channel (CFTR) works well enough that there are no medical problems. However, if the CF gene inherited from both parents has mutations, then the salt channel does not work at all (or only works a tiny bit) and then, there are medical consequences related to the thick mucus. People with CF commonly have lung, digestive, nutritional and reproductive problems.
Although we are constantly searching for a permanent solution to CF, there is no cure….yet! But, with appropriate therapy, it is possible to slow the progression of disease and control complications from CF.
To make the diagnosis of CF there must be a combination of certain typical clinical symptoms or medical conditions plus tests that demonstrate an abnormality in the amount of salt in sweat or genetic testing that shows the presence of 2 mutations in the CF gene. Since 2008, in Ontario, all newborn babies are screened for CF.
Some examples of typical CF symptoms are: failure to gain weight in spite of a good appetite; frequent greasy, loose bowel movements; bowel blockage at birth (meconium ileus); chronic cough; frequent lung infections; nasal polyps; chronic sinusitis; male infertility.
Individuals that have the characteristic symptoms, would then have a blood genetic testing or a sweat test.
More information can be found here: https://www.cff.org/What-is-CF/Diagnosed-With-Cystic-Fibrosis/
The CF gene was first discovered in 1989 by a team of investigators at the Hospital for Sick Children. It took a further 2 years for scientists to discover exactly what the CF gene is for. The CF gene contains the instructions on how to build a channel that sits in the membrane of some cells in the body. This channel is a protein and is called CFTR (cystic fibrosis transmembrane regulator) and it functions as a pore that chloride and bicarbonate can move through. CFTR also influences other channels and is critical in determining the amount of water in mucus.
Mutations in the CF gene are mistakes in the DNA code that cause dysfunction in CFTR (so that the salt channel does not work properly). During the time that the CF gene was discovered, about 10 different mutations were described. We now know that there are more than 2,000 mutations in the CF gene. Some of these mistakes can be more severe than others. If you were reading instructions and there was an entire page missing, that would be a severe mistake and it would be difficult to follow the instructions. However, if the mistake was a misspelled word, you could still probably follow the instructions and this would be a mild mistake. In this way, some of the 2000 mutations are severe and some are mild and this has a consequence on the severity of symptoms.
The most common CF mutation: Delta F508 (or F508del)
The most common mutation causing CF is called delta F508 – a deletion of an amino acid called phenylalanine at position 508 in the gene. This results in CFTR not being able to fold into the proper shape. The body recognizes this abnormality and breaks down the protein after it is made so that CFTR does not reach its correct position in the cell membrane. Thus, the delta F508 mutation is a severe mutation. Half of the people with CF in Canada carry 2 copies of the deltaF508 mutation (getting 1 copy from each parent) and 40% have one copy of deltaF508 and a different second mutation.
Scientists have divided the 2000 different mutations in the CF gene into categories based on their impact on the CFTR protein.
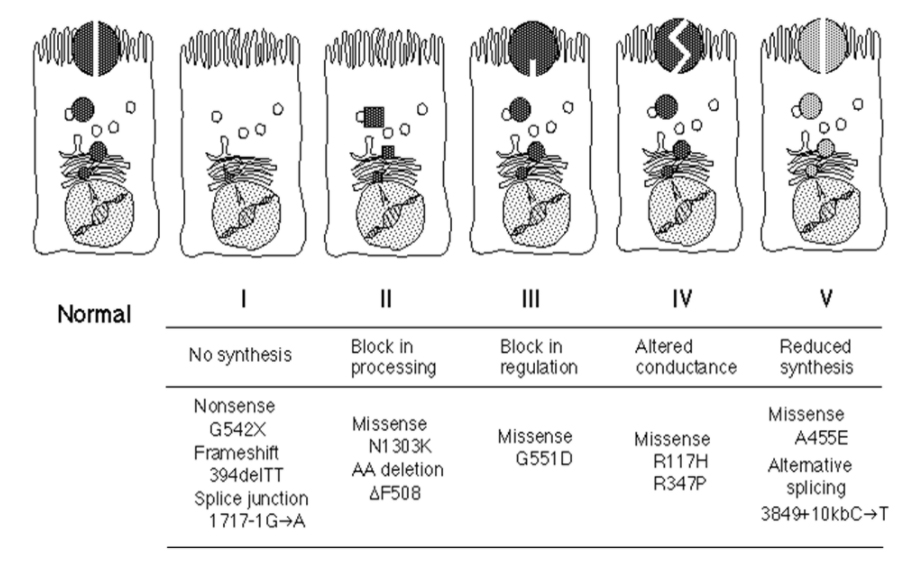
Classification of CF Mutations
Type I mutations are so severe that no normal CFTR protein is made at all. An example of Type 1 mutations are stop mutations. Normally, the DNA contains in structions to say when the CFTR protein is complete. If there is a stop mutation, it is as if the last few pages of the instructions are missing so you are left with a CFTR protein that is only partially completed and thus does not work as a salt channel. The more common of these mutations are the G542X and W1282X mutations.
Type II mutations are mistakes that cause a problem with processing the CFTR protein. The CFTR protein is not made correctly so the body breaks down the protein before it can reach the cell surface. Delta F508 is the most common Type 2 mutation. Type 2 mutations are considered severe mutations.
Type III mutations are mistakes that mean that, although the salt channel is made correctly and gets to the correct position in the membrane of the cell, it does not work. The G551D mutation is an example of a Type 3 mutation and Type 3 mutations are also severe mutations.
Type IV mutations are considered mild mutations because the CFTR protein is in the correct location on the cell membrane and it does work to move salt but it doesn’t function properly. This is also called a conductance problem. The R117H mutation is the most common Type 4 mutation.
Type V mutations are also mild mutations where the number of salt channels is reduced but each individual channel functions normally. These are very rare mutations and include 3849+10kbC->T and A455E.
The CFTR2 website has a lot of good information about CF mutations.
How does a problem in CFTR cause problems in the lungs and digestive tract?
The CFTR protein sits in the cell membrane and acts as a channel for both chloride and bicarbonate, moving these salt ions out of the cell in the parts of the body where mucus is made. The movement of chloride helps control the movement of water into the mucus. When CFTR protein is absent or not working properly, the mucous becomes dehydrated, thick, and does not move easily.
The surface of the airways in the lungs is covered by a thin layer of mucus. This mucus traps bacteria that are breathed into the lung. The mucus is moved along the airways by little hairs on the surface of the cells called “cilia”. The job of the cilia is to sweep the mucus out of the lung. It is much harder for the thick mucus in CF to be moved out of the lung by the cilia. In order to try and get rid of the mucus, you need to cough.
The nutrients and lack of oxygen in the mucus create the perfect environment for certain bacteria to thrive in your lungs. Thus, in the lungs of people with CF, bacteria start to grow in the mucus inside the airways. The immune system of the body is alerted and sends cells to try and kill the bacteria. Over time these immune cells cause inflammation damaging the lungs and causing the cells to make more mucus. This creates a vicious cycle of mucus plugging of airways, infection, inflammation, and more mucus plugging which blocks the airways. The dilated (or stretched) airways with thickened, scarred walls create the characteristic lung changes of CF called bronchiectasis.
In the digestive tract, thick mucus can form a plug blocking the bowels (in infants, this is meconium ileus and in children or adults, this is distal intestinal obstruction syndrome). Thick mucus blocks the ducts in the pancreas preventing pancreatic enzymes from being released to digest food (pancreatic insufficiency) and can also plug the ducts in the liver leading to scarring (biliary cirrhosis).
Cystic Fibrosis can have ramifications throughout your body. Continue exploring this section to learn more about the various systems and how they might be affected.
Basic Facts
Cystic fibrosis (CF) is a common genetic disease that is inherited from your biological parents. It is the most common genetic disease in Caucasians; approximately 1 in every 3,600 children born in Canada has CF.
Genetic illnesses occur because of abnormalities in our genes.
All the cells in our body contain DNA. Simply put, our DNA is the instructions on how to build us. If you think of our DNA like a library, then all these instructions are divided into “books” – called chromosomes and within each book, there are “chapters’, called genes. Each gene contains detailed instructions on how to build one part of our body.
Just like there can be mistakes in written instructions, there can be mistakes in our DNA. A mistake in our DNA is called a mutation.
The CF gene is the instructions on how to build a channel (or pore) in our cells that allows salts (chloride and bicarbonate) to move into and out of the cell. This salt channel is called CFTR, the cystic fibrosis transmembrane regulator. A mutation in the CF gene means that there is a mistake in the instructions on how to build this salt channel so it does not work properly.
CFTR is important in regulating the amount of water in the mucus in our bodies – for example in the lungs, sinuses, and digestive tract. If CFTR does not work properly, the mucus is abnormally thick and sticky.
We inherit a copy of our genes from our parents – one copy from our father and one from our mother. If only one copy of the CF gene with a mistake (mutation) is inherited, then the individual is a “carrier” of the CF gene. This is because they carry the mutation (and can pass it on to their children), but because the copy of the gene inherited from the other parent is normal, the salt channel (CFTR) works well enough that there are no medical problems. However, if the CF gene inherited from both parents has mutations, then the salt channel does not work at all (or only works a tiny bit) and then, there are medical consequences related to the thick mucus. People with CF commonly have lung, digestive, nutritional and reproductive problems.
Although we are constantly searching for a permanent solution to CF, there is no cure….yet! But, with appropriate therapy, it is possible to slow the progression of disease and control complications from CF.
To make the diagnosis of CF there must be a combination of certain typical clinical symptoms or medical conditions plus tests that demonstrate an abnormality in the amount of salt in sweat or genetic testing that shows the presence of 2 mutations in the CF gene. Since 2008, in Ontario, all newborn babies are screened for CF.
Some examples of typical CF symptoms are: failure to gain weight in spite of a good appetite; frequent greasy, loose bowel movements; bowel blockage at birth (meconium ileus); chronic cough; frequent lung infections; nasal polyps; chronic sinusitis; male infertility.
Individuals that have the characteristic symptoms, would then have a blood genetic testing or a sweat test.
More information can be found here: https://www.cff.org/What-is-CF/Diagnosed-With-Cystic-Fibrosis/
The CF gene was first discovered in 1989 by a team of investigators at the Hospital for Sick Children. It took a further 2 years for scientists to discover exactly what the CF gene is for. The CF gene contains the instructions on how to build a channel that sits in the membrane of some cells in the body. This channel is a protein and is called CFTR (cystic fibrosis transmembrane regulator) and it functions as a pore that chloride and bicarbonate can move through. CFTR also influences other channels and is critical in determining the amount of water in mucus.
Mutations in the CF gene are mistakes in the DNA code that cause dysfunction in CFTR (so that the salt channel does not work properly). During the time that the CF gene was discovered, about 10 different mutations were described. We now know that there are more than 2,000 mutations in the CF gene. Some of these mistakes can be more severe than others. If you were reading instructions and there was an entire page missing, that would be a severe mistake and it would be difficult to follow the instructions. However, if the mistake was a misspelled word, you could still probably follow the instructions and this would be a mild mistake. In this way, some of the 2000 mutations are severe and some are mild and this has a consequence on the severity of symptoms.
The most common CF mutation: Delta F508 (or F508del)
The most common mutation causing CF is called delta F508 – a deletion of an amino acid called phenylalanine at position 508 in the gene. This results in CFTR not being able to fold into the proper shape. The body recognizes this abnormality and breaks down the protein after it is made so that CFTR does not reach its correct position in the cell membrane. Thus, the delta F508 mutation is a severe mutation. Half of the people with CF in Canada carry 2 copies of the deltaF508 mutation (getting 1 copy from each parent) and 40% have one copy of deltaF508 and a different second mutation.
Scientists have divided the 2000 different mutations in the CF gene into categories based on their impact on the CFTR protein.
Classification of CF Mutations
Type I mutations are so severe that no normal CFTR protein is made at all. An example of Type 1 mutations are stop mutations. Normally, the DNA contains in structions to say when the CFTR protein is complete. If there is a stop mutation, it is as if the last few pages of the instructions are missing so you are left with a CFTR protein that is only partially completed and thus does not work as a salt channel. The more common of these mutations are the G542X and W1282X mutations.
Type II mutations are mistakes that cause a problem with processing the CFTR protein. The CFTR protein is not made correctly so the body breaks down the protein before it can reach the cell surface. Delta F508 is the most common Type 2 mutation. Type 2 mutations are considered severe mutations.
Type III mutations are mistakes that mean that, although the salt channel is made correctly and gets to the correct position in the membrane of the cell, it does not work. The G551D mutation is an example of a Type 3 mutation and Type 3 mutations are also severe mutations.
Type IV mutations are considered mild mutations because the CFTR protein is in the correct location on the cell membrane and it does work to move salt but it doesn’t function properly. This is also called a conductance problem. The R117H mutation is the most common Type 4 mutation.
Type V mutations are also mild mutations where the number of salt channels is reduced but each individual channel functions normally. These are very rare mutations and include 3849+10kbC->T and A455E.
The CFTR2 website has a lot of good information about CF mutations.
How does a problem in CFTR cause problems in the lungs and digestive tract?
The CFTR protein sits in the cell membrane and acts as a channel for both chloride and bicarbonate, moving these salt ions out of the cell in the parts of the body where mucus is made. The movement of chloride helps control the movement of water into the mucus. When CFTR protein is absent or not working properly, the mucous becomes dehydrated, thick, and does not move easily.
The surface of the airways in the lungs is covered by a thin layer of mucus. This mucus traps bacteria that are breathed into the lung. The mucus is moved along the airways by little hairs on the surface of the cells called “cilia”. The job of the cilia is to sweep the mucus out of the lung. It is much harder for the thick mucus in CF to be moved out of the lung by the cilia. In order to try and get rid of the mucus, you need to cough.
The nutrients and lack of oxygen in the mucus create the perfect environment for certain bacteria to thrive in your lungs. Thus, in the lungs of people with CF, bacteria start to grow in the mucus inside the airways. The immune system of the body is alerted and sends cells to try and kill the bacteria. Over time these immune cells cause inflammation damaging the lungs and causing the cells to make more mucus. This creates a vicious cycle of mucus plugging of airways, infection, inflammation, and more mucus plugging which blocks the airways. The dilated (or stretched) airways with thickened, scarred walls create the characteristic lung changes of CF called bronchiectasis.
In the digestive tract, thick mucus can form a plug blocking the bowels (in infants, this is meconium ileus and in children or adults, this is distal intestinal obstruction syndrome). Thick mucus blocks the ducts in the pancreas preventing pancreatic enzymes from being released to digest food (pancreatic insufficiency) and can also plug the ducts in the liver leading to scarring (biliary cirrhosis).